
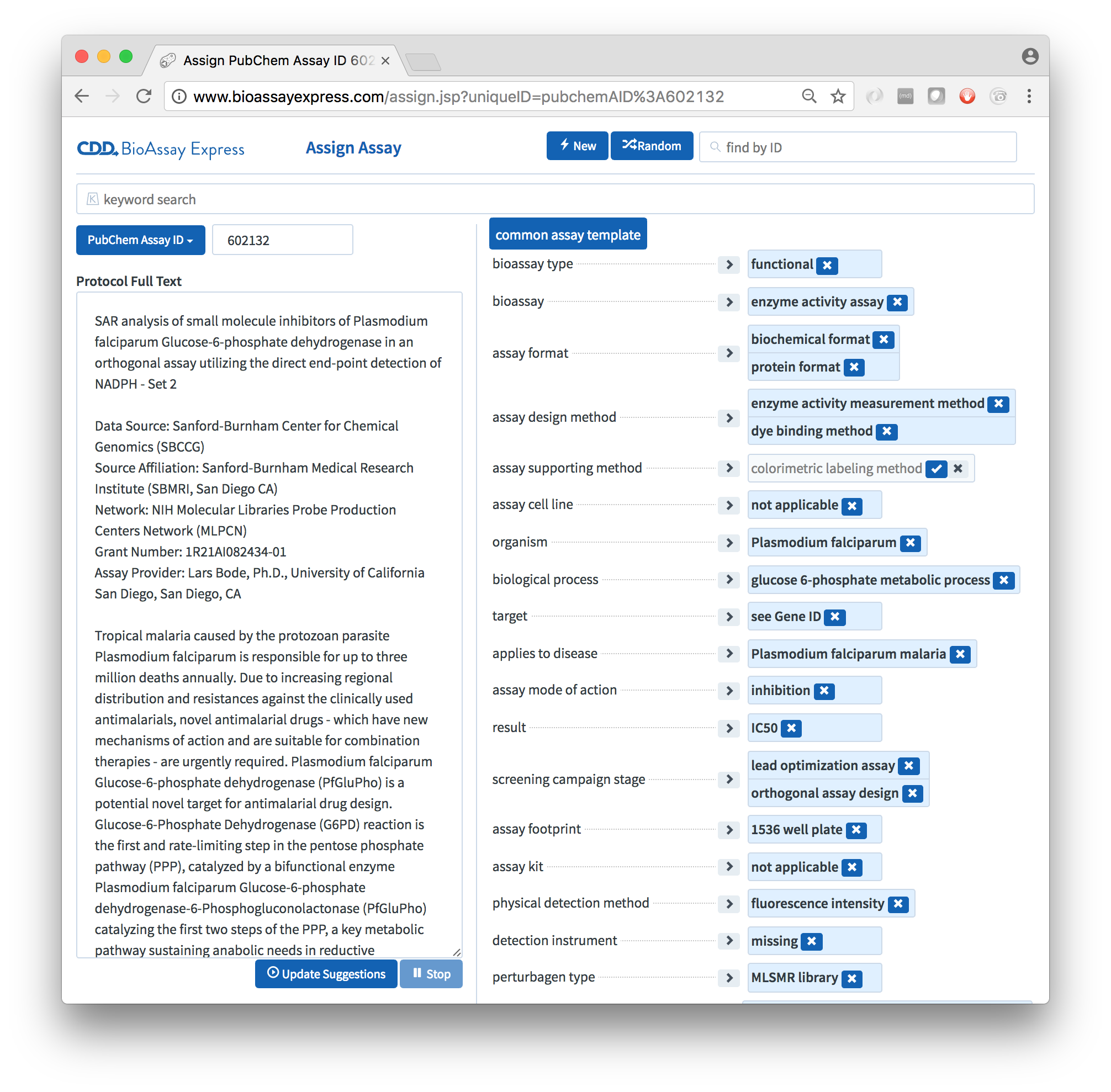
The BioAssay Express is a web-based tool for annotating bioassay protocols using semantic web terms. Curation involves a human operator, assisted by machine learning and an ergonomic UI/UX. Each assay is annotated once, after which it becomes fully machine readable: assays can be searched, sorted, clustered and analyzed without ever having to read through the original text. This approach is suitable for institutions with assay protocols stored in legacy formats, for creating new content as an electronic lab notebook, and for exploring structure-activity relationships within large volumes of public data.
One of the key activities in early stage drug discovery is the screening of potential chemical entities (either small molecules or larger biological fragments) to test for activity against various targets (which include specific proteins for diseases or biological processes, whole cell tests, ADME/toxicology, etc.). Each screening configuration is a distinct scientific experiment, and even when two scientists are measuring the same property, no two experiments are ever identical: and all too often, the nuances are important.
The problem that the BioAssay Express was created to solve is that current methods for recording bioassay protocols are very far behind other aspects of drug discovery. The standard practice for assay protocols is to document them using scientific English, which is effective for communication between screening biology experts, but it becomes a rapidly escalating challenge to manage large collections of protocol descriptions. Many companies and institutions have developed in-house solutions for collating their growing assay collections, typically using bespoke user interfaces to encourage scientists to select labels from lists of controlled vocabulary.
Common issues with the status quo of assay management include:
| text assays are only searchable by keywords, which have a high failure rate (false positives & false negatives abound) |
| marking up assays with controlled vocabulary helps, but the terms are not interchangeable between institutions, providing no value for collaboration or externalization |
| controlled vocabulary terms are often poorly defined and optional, resulting in sporadic coverage or imperfect understanding |
| compliance with documentation policies is difficult to measure or enforce |
| text descriptions are time consuming to review, and it is easy to make mistakes |
| reproducibility suffers from the absence of rigorously defined annotation standards |
| institutional knowledge is frequently lost: assays are often repeated unnecessarily |
| many scientists dislike or have difficulty using scientific English to document everything they do |
| there are no easy ways to peruse similar assays, or analyze previous work, or compare experiments with other institutions or public data |
| mergers and acquisitions are an information systems nightmare |
The BioAssay Express addresses all of these issues by using semantic standards, allowing scientists to do a one-time curation of each of their assays, with minimum fuss.
The overall objective of the project is to make assay informatics into a standard practice within the drug discovery field: this approach has been overwhelmingly successful for cheminformatics and bioinformatics, and there are numerous ways in which this can aid projects:
| query with precision: find assays immediately and reliably, without missing anything or digging through superfluous matches |
| tag assays with labels - it becomes easy to see what belongs to each category |
| standardized ontologies maximize data compatibility, both for public datasets and private collaborations |
| combining datasets from two different institutions becomes simple, as long as they are both using semantic annotations: one less reason to be terrified of mergers and acquisitions |
| universal vocabulary makes it much easier to train scientists to use them correctly |
| the underlying public ontologies have been very carefully vetted by domain experts, and the implicit hierarchy, labels and descriptions provide valuable extra metadata |
| science evolves, and so do the ontologies: new terms to describe new biology concepts (e.g. newly druggable targets) can be easily incorporated at regular intervals |
| semantic web terms can be assigned multiple labels, meaning that translation to other languages is a cosmetic change |
| annotation during registration makes completeness easy to monitor: missing content can be filled in later, or detected with simple reports |
| document the key concepts using semantic annotations, and use full text to describe the rest |
| publicly annotated data can be imported and mixed with private content, to increase domain knowledge and improve structure-activity relationship predictions |
| related assays can be discovered with ease, which can promote internal collaboration, as well as reduce wasteful repetition of experiments |
| highly structured descriptions of assays allow analysis techniques that were previously impractical, e.g. clustering, aggregating, filtering and combining with compound measurements |
| cloning an assay is easy, and much less error prone: duplicate the terms, then change only those which are different |
| template customization allows the creation of business rules for biologists, which are much easier to understand - as well as enforce - than text |
| the choice between public vs. private applies to assays, templates and ontologies: mixing public and private content behind a firewall allows choice of tradeoff between secrecy and seemless collaboration |
| annotate each assay once, gain value from it forever |
The semantic web provides a great starting point for universally understood descriptive labels that can be applied to any concept, including biology. Because of the efforts of projects such as BioAssay Ontology Cell Line Ontology, Gene Ontology, Drug Target Ontology and many others, there are enough well defined terms in the public domain to provide the vocabulary necessary to describe most all of the details involved in a screening experiment. These semantic web terms are highly appropriate for documenting laboratory procedures because they are at their core a machine readable technology, whereby every annotation is precisely and unambiguously defined. Additionally, the annotations can easily be presented to humans in a meaningful way, which means that it is possible to achieve the best of both worlds: perfect indexing and high readability.
In spite of its favorable characteristics and expressive power, the semantic web is only as good as its data. Using the raw tools that are available for annotation is rather like being handed a dictionary and being expected to write a novel. For practical purposes, content creation requires further guidelines for how to use the terms (which can be thought of as the grammar), as well as an efficient and easy to learn user interface, and ideally a collection of prior examples.
In order to reduce the massive degrees of freedom that the raw semantic web allows, the BioAssay Express project uses assay templates to define assignment categories, and the applicable terms within each one (this underlying technology is open source, and can be found on GitHub, and has also been described in the literature).
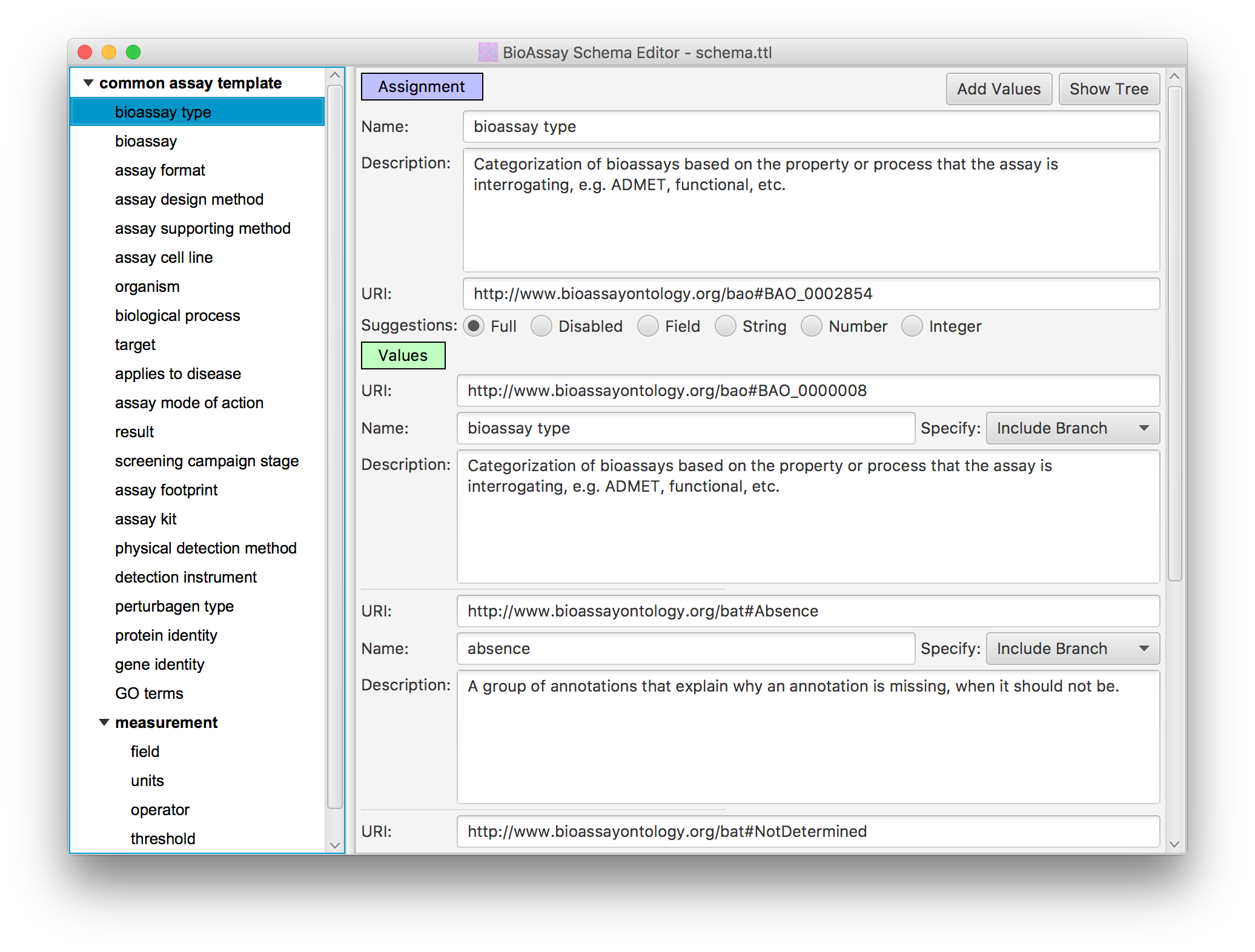
The default template for public data is called the Common Assay Template, which consists of a couple dozen assignment categories, which are designed to capture most of the summary characteristics of a bioassay protocol, most of the time. While it lacks the depth needed to replace the detailed text description, it provides enough information about the assays to perform sophisticated searching and analysis, to an extent that is not possible with alternative assay management systems.
In practice, any number of assay templates can created, and some of them can be very detailed, and delve into the finer details of a protocol. The available terms that the templates use are drawn from a list of ontologies, most of which are public and maintained by independent stakeholders. The public ontologies can be supplemented by any number of private extensions, allowing an unlimited amount of template customization.
Since most of the world's assay data is stored in the form of text descriptions, this means that most public data, or legacy content from within an institution, requires some amount of curation. While it is possible to devise a mapping system to import assays that were marked up with a custom designed controlled vocabulary, these typically cover only a handful of the fields, if they are available at all. Recognizing that the legacy curation process is one of the largest barriers to entry, the BioAssay Express devotes a significant amount of energy to making the curation as painless as possible.
The underlying principle is that both extremes of automation or lack thereof are impractical:
The BioAssay Express splits the difference and makes heavy use of machine learning to minimize the amount of time and expertise required to curate each assay, rather than to eliminate the human curator altogether. This hybrid approach has been used to reduce the curation time by an order of magnitude or more (which we have described in the literature, as well as being awarded a patent for).
The machine learning support is divided into two steps: the first part involves analyzing all of the available assay text using natural language processing, to partition the sentence structure into parts-of-speech blocks, which are fed into a Bayesian model for each and every annotation term that has been encountered thus far. This means that a fresh database has essentially no idea how to make recommendations for annotating an assay, but as the documents start to accumulate, correlations between text and annotations begin to resolve themselves. The association between the text and the ability to predict which annotations are correct is quite fuzzy, but this is appropriate for the use case: the objective is to guess the right terms most of the time, so that the user can scan through the suggestions and quickly confirm those which are correct. The penalty for failure manifests in the form of wasting a scientist's valuable time, which is certainly to be avoided, but this is far less severe than creating false data.
The second part of the machine learning is the building of correlation models, which also use the Bayesian method, for observing when combinations of terms are more or less likely to be used together. This is effective because assay protocol annotations are frequently non-orthogonal (e.g. disease and protein target are strongly interrelated, as are detection instrument and physical detection method). These correlation models can be used to refine the quality of suggestions as the annotation process continues.
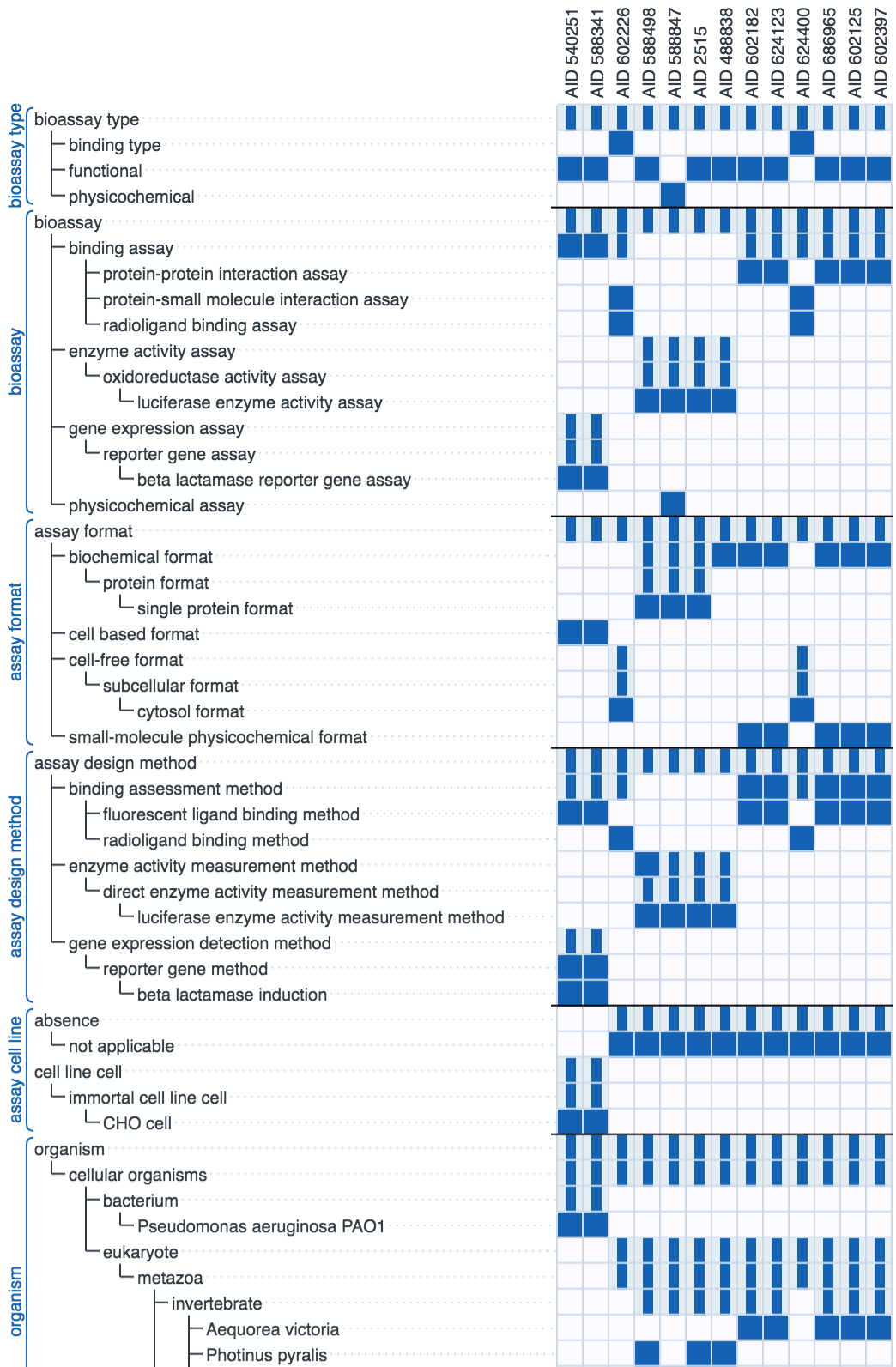
The diagram above shows the recall rate for a test set of previously curated assays: each row shows the highest rated annotation suggestions from left to right. Each correct suggestion is marked as a dark blue dot. The graphic simulates the human/machine hybrid approach by assuming that each correct suggestion is approved by a human operator, which allows it to be added to the secondary correlation model to improve the remaining suggestions. In all cases the majority of the annotations are correctly guessed early on in the sequence, although it can be seen that there are a handful of annotations for which their models perform very poorly, which is why a human presence is always necessary.
In short, the machine learning technology is useful for accelerating bulk annotation of a series of assays with a fair bit of commonality, which is often the case for legacy data. The technique's strength is its ability to learn from literally anything, but it lacks any kind of predictive ability for unfamiliar content, and can provide relatively little assistance for assays that are significantly different from what has been curated already.
The components that make up the BioAssay Express were developed by testing the curation interface on a collection of public data, using the Common Assay Template. While there are millions of bioassay protocols that can be found in public databases, and untold more that are still locked away within the primary scientific literature, there are relatively few public sources that provide detailed text descriptions for assay protocols. One of the most valuable resources is a subset of the information stored within the PubChem database deposited by the Molecular Libraries Program.
During the summer of 2016, a small team at Collaborative Drug Discovery curated just over 3,500 of these assays, using the BioAssay Express project, while simultaneously refining the Common Assay Template, iteratively improving what was initially a relatively crude user interface, requesting new ontology terms and tweaking the machine learning support. In spite of the work-in-progress nature of the project, the typical curation time per assay was measured in minutes.
The resulting annotation content has been made publicly available, and can be accessed using the main website at www.bioassayexpress.com. While the number of assays is a small fraction of the entire PubChem collection, it does represent a valuable resource, both for real world use, and for designing and testing new technologies for making use of well annotated assay data. This resource can be used by itself, or it can be combined with private data.
It is also possible to join in on the annotation process: the beta website (beta.bioassayexpress.com) allows anyone to authenticate themselves and submit their own annotations for assays that have been selected for inclusion within the dataset. Scientists are invited to join in and crowd source the curation of this increasing valuable resource.
Curation of legacy data is the single largest bottleneck, but the quantity is finite. The BioAssay Express is designed to be equally applicable to the curation of new data, as it is generated. Typically when writing up an experiment that is imminent or complete, there is no convenient text description to help the machine learning algorithms, but it is often very convenient to simply hunt through the existing data to find the most similar experiment, and clone it. The cloned record can be quickly adjusted as necessary (e.g. just changing the target and protein for an otherwise identical screening run).
The web-based user interface is designed so that selection of values for each of the assignment categories is fast - in most cases, more convenient than actually writing the text. The data entry page contains a lot of keyboard shortcuts and convenient ways to navigate the available options, in addition to the fact that the machine learning algorithms can provide some level of support, even without any text description, once the first few annotations have been asserted.
Once an assay protocol has been annotated with semantic web terms, the range and diversity of algorithms that can be used to organize, filter, sort, group or study them is almost limitless, and all of them can be carried out without requiring a human expert to go back to the original data and read through it.

One of the most effective ways to explore the assay collection is via the Explore interface, which is essentially a filter: a series of selection steps are applied, each of which reduces the qualifying assays - starting from everything, and eventually winnowing down to a selected subset that contains everything of interest. Each filtering step involves picking an assignment category, then selecting terms (sometimes a whole branch at once). This can be used to quickly look at one category to examine the diversity of annotations, or it can be used to very specifically refine a set of assays, which can be examined or used for a subsequent purpose.
The Explore interface is dynamic and responds immediately whenever a term is clicked upon, which makes it effective for exploring assays when one is not sure what to expect. It also provides the ability to filter based on keywords (or numeric ranges), which is less precise than using semantic web terms, but can be bluntly effective.
The annotations that adorn the assays can be treated like fingerprints, each of which is either present or absent. We can look the the adjacent discipline of cheminformatics for ideas on how to treat these: for example, it is straightforward to compute a Tanimoto similarity coefficient for two assays. The Search feature performs a comparison method that is conceptually similar, with some modifications to make use of the assignment categories and the hierarchical nature of the annotation terms. Searching for assays involves specifying a set of annotations and starting the search, which retrieves a list of assays ranked in decreasing order of similarity. For convenience, it is possible to pick an existing assay and initiate a search using its own annotations as the starting point, i.e. the "self" will return a similarity of 100%.
A variety of summary pages are available, e.g. the assay counts for each kind of annotation, contributions from various sources, issues and inconsistencies, etc. This functionality is readily adapted in order to keep track of the health of the database.
The BioAssay Express is a commercial product, designed and built by Collaborative Drug Discovery, Inc.. The public-facing instance (at www.bioassayexpress.com), and a large amount of assay curation based on the original PubChem data, was supported in part by a small business grant and is made available as a free service.
For inquiries about running the product privately, contact us.
The user interface for BioAssay Express is purely web-based and runs on any modern browser. The JavaScript codebase is written using cross-compiled TypeScript. The middleware is written in Java, using the J2EE framework, and is tested and deployed from Apache Tomcat. Natural language processing is done with Apache OpenNLP, semantic web processing with Apache Jena, and cheminformatics with the Chemical Development Kit. The back-end database is the NoSQL MongoDB.